8. Differential expression (with EBSeq)¶
Note
You can also start with the data on snap-1025bf17; mount it as /data and do:
cd /mnt
gunzip -c /data/nematostella.fa.gz > ./nematostella.fa
mkdir ebseq
cd ebseq
rsem-generate-data-matrix /data/[0-9].fq.genes.results /data/10.fq.genes.results > 0-vs-6-hour.matrix
Note
If all you have is a genes.results file, you can recover the *.fq.genes.results files by doing the following:
python /usr/local/share/eel-pond/unpack-genes-matrix.py genes.results
Now run rsem-generate-data-matrix to put together the columns you’re
interested in for the pairwise comparison.
Go to /mnt and make a new directory:
cd /mnt
mkdir ebseq
cd ebseq
cp ../rsem/0-vs-6-hour.matrix .
Next, run EBSeq:
rsem-run-ebseq 0-vs-6-hour.matrix 5,5 0-vs-6-hour.changed
Here, the .matrix file contains 2 conditions, each with 5 replicates; if you had two replicates, you would call rsem-run-ebseq with 2,2.
The EBSeq output will be in ‘0-vs-6-hour.changed’. Read the docs to understand what’s in the output file – you’re most interested in the PPDE (posterior probability that a transcript is differentially expressed) and the PostFC (posterior fold change) columns, columns 4 and 5.
Finally, let’s extract differentially expressed genes, and combine them with the annotations in your transcripts file. :
python /usr/local/share/eel-pond/extract-and-annotate-changed.py 0-vs-6-hour.changed /mnt/nematostella.fa 0-vs-6-hour.changed.csv
This will produce a file containing many rows, each with 5 columns: each row is a transcript family, and the columns are the probability of that transcript family being differentially expressed (according to EBSeq), the posterior fold change (calculated by EBSeq), the real fold change (EBSeq), the transcript family name, and any annotations that have been assigned to that transcript family.
This file can be opened directly in Excel or most any spreadsheet program.
To visualize the distribution of gene expression in the two conditions you can do:
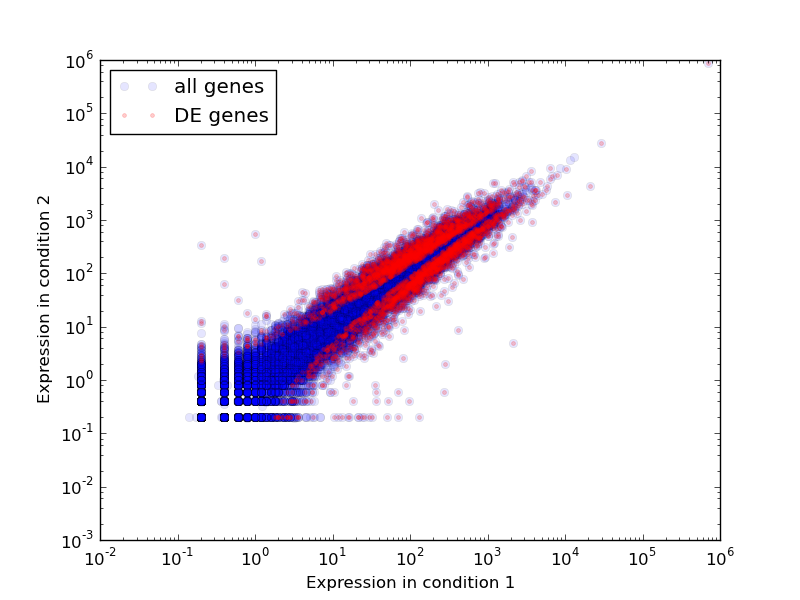
python /usr/local/share/eel-pond/plot-expression.py 0-vs-6-hour.matrix 5,5 0-vs-6-hour.changed.csv
This will produce a .PNG image showing all of the genes’ expression levels in condition 1 against their levels in condition 2, and will show in a separate color those genes that are differentially expressed. Running it on the demo data set will produce an image as below
...and that’s all, folks!
LICENSE: This documentation and all textual/graphic site content is licensed under the Creative Commons - 0 License (CC0) -- fork @ github.
comments powered by Disqus